Jun 29, 2023 · 14 min read
HyenaDNA: learning from DNA with 1 Million token context

Image generated by Adobe Firefly with prompt, "cute Hyena with DNA sequences".
We're excited to introduce HyenaDNA, a long-range genomic foundation model with context lengths of up to 1 million tokens at single nucleotide resolution!
HyenaDNA is pretrained on the human reference genome, and sets new SotA on 23 downstream tasks including predicting regulatory elements, chromatin profiles, and species classification. We also explore what new capabilities open up with long context in genomics, including in-context learning with soft prompt tuneable tokens and instruction fine-tuning.
Resources
arxiv
colab
github
huggingface (ckpts)
youtube (talk)
Long context for DNA
Long context models are all the rage these days. OpenAI stated a goal to reach 1M tokens, while Magic announced they reached 5M tokens for code. We’re all for it, the more minds working on it, the more progress and amazing use cases! But while most have focused on natural language and code (in the long context arms race), we noticed no one was paying much attention to a field that is inherently made of ultralong sequences: biology.
In particular, the field with the longest sequences is arguably genomics, which is the study of all genetic material present in an organism. It includes the analysis of its function, structure, and evolution. To give a sense of the scale, the human genome (an entire set of DNA) has 3.2 billion nucleotides, which are the building blocks of DNA and can be seen as “characters” in the sequence.
Recently, Hyena, a convolutional LLM, showed it could match attention in quality with lower compute and time complexity, enabling much longer sequences in associative recall tasks. (check out our other blogs on Hyena: blog 1, blog 2, blog 3)
But could Hyena work on genomics? What new capabilities could a long context genomic foundation model (FM) enable?
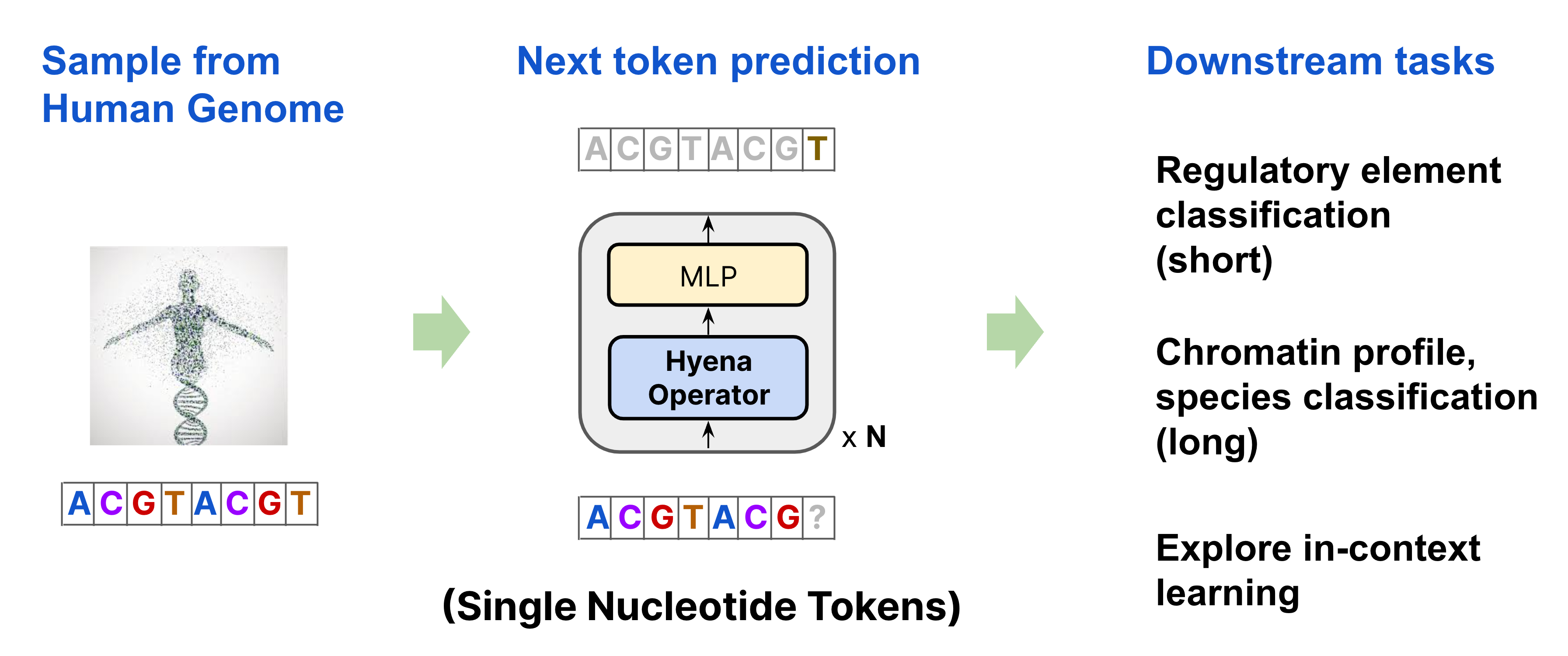
To pretrain HyenaDNA, we randomly sample a sequence from the Human Reference Genome and learn to predict the next nucleotide. Our architecture is a simple stack of Hyena operators, with a single character tokenizer, and primary vocabulary of 4 (A, C, T, G). We then apply the pretrained model on over 28 downstream tasks, as well as explore the first use of in-context learning in genomics.
HyenaDNA: combining Hyena and genomics
Turns out, the answer is yes! We're *excited* to introduce HyenaDNA, a genomic foundation model pretrained on sequences of up to 1 million tokens long at single nucleotide resolution. We show that as one increases context length, we can reach better performance measured in improved perplexity. HyenaDNA trains up to 160x faster than Transformers with FlashAttention, uses a single-character tokenizer, and has global context at each layer. We apply HyenaDNA on 28 genomic tasks (SotA on 23), using far fewer parameters than previous genomic models, with examples that can fit on colab.
We'll definitely dive into the experiments later, as well as the model details of HyenaDNA. But first, we want to share why we think genomics is an incredibly fascinating (and potentially underexplored) domain for deep learning research.
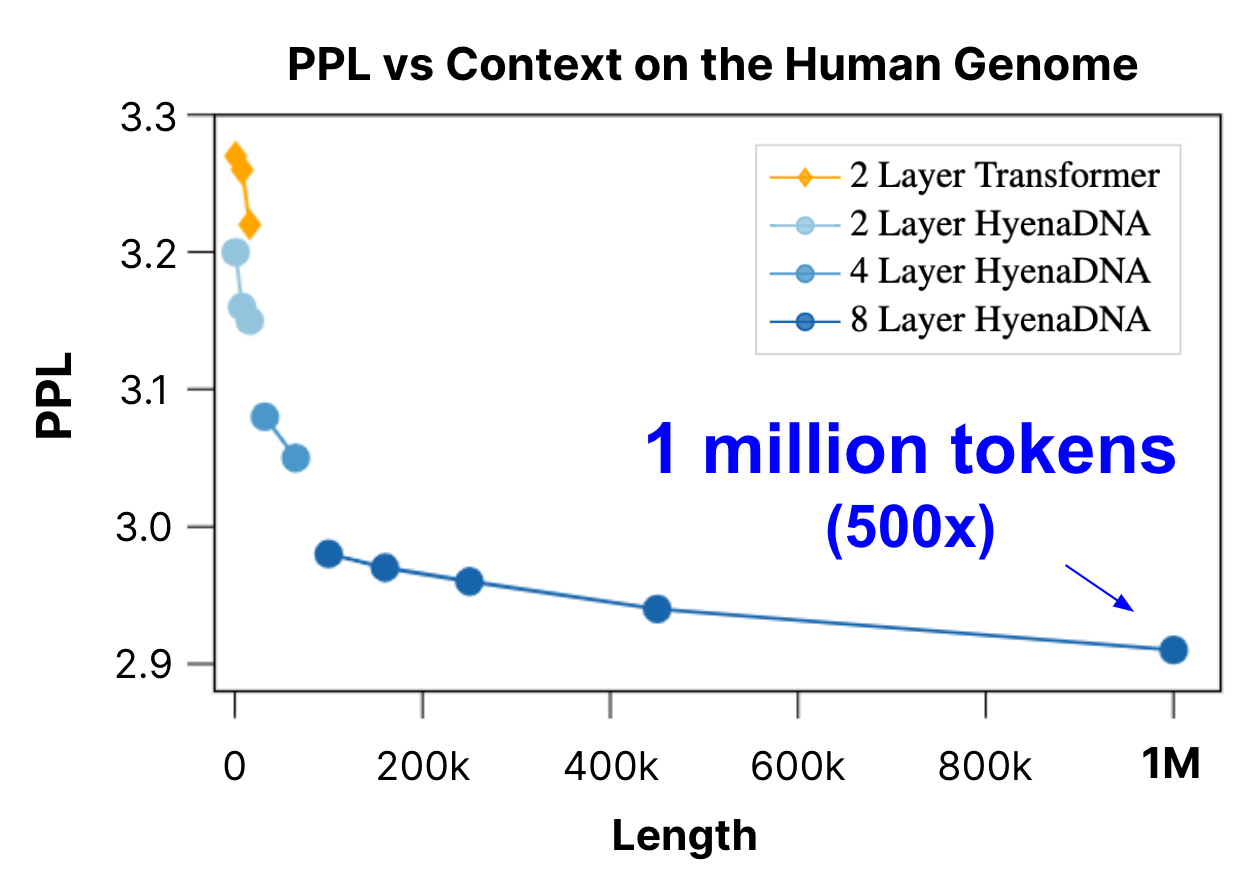
We pretrain a family of HyenaDNA models across model size and sequence length. We observe that as we increase context length, we're able to achieve better perplexity (improved next token accuracy). For comparison, we also benchmark against a Transformer (up to 16k tokens). At 1 million tokens, HyenaDNA is up to 500x longer than previous genomic FMs.
Why focus on genomics?
As fun as increasing context length benchmarks is, the potential impact of improving context lengths and expressivity in genomics is what’s compelling to us. Although the human genome has been mapped, which tells us the order of nucleotides in the sequence, how the sequences space map to function, traits, disease, and many other questions, remain an ongoing research effort. Cracking the genetic code would help us better understand disease (genetic disorders), and enable breakthroughs in drug discovery through modeling gene expression (and identifying drug targets). Imagine being able to prompt ChatGPT with an entire human genome - wouldn't it be neat to query and ask questions about likely diseases, predict drug reactions, or guide treatment options based on your specific genetic code? (in the future...)
Previous Genomic FMs
There has been amazing work applying foundation models (aka LLMs) to genomics [DNABERT, Nucleotide Transformer, GenSLMs, GENA-LM], modeling DNA as the “language” of life. Unfortunately, these works have been limited by the quadratic scaling of attention in Transformers, and thus far have typically used context lengths of between 512 - 4k tokens, depending on dense or sparse attention. That’s less than 0.001% of the length of the human genome. (We also noticed that compared to protein models, which have fairly "short-range" sequences, genomics is far less established.)
Equally important and challenging is the need for high resolution. Virtually all genomic FMs rely on tokenizers to aggregate meaningful DNA units or “words”, or use fixed k-mers, which have fixed “words” of size k that overlap with each other. These “hacks” help lengthen context as well. However, the existence of single nucleotide polymorphisms (SNPs, pronounced as “snips”), show that single DNA character changes can completely alter genes, protein function, or cause a cell to enter a disease state.
That means both long context and single nucleotide resolution is critical in genomics! In contrast, for natural language, a single character or word change in a chapter doesn’t change the semantics really. There has been work with single character tokenizer, but they've struggled with both the longer sequences, and underperforming BPE or subword tokenizers [Clark et al., Tay et al.]. We hope to challenge that paradigm.
What are genomic FMs trying to learn?
Let’s provide some intuition about what we’re trying to learn from DNA. For that, we need a little bit of a biology primer (from a 30k foot view). Feel free to skip this section if you know some basic biology already.
Every cell in your body has a complete copy of your entire DNA, encoding all the instructions needed to build proteins and biological “products” to sustain life. But cells can differentiate - a brain cell is different from a heart cell, is different from a liver cell, etc. Same DNA instructions, but different cells, what gives?
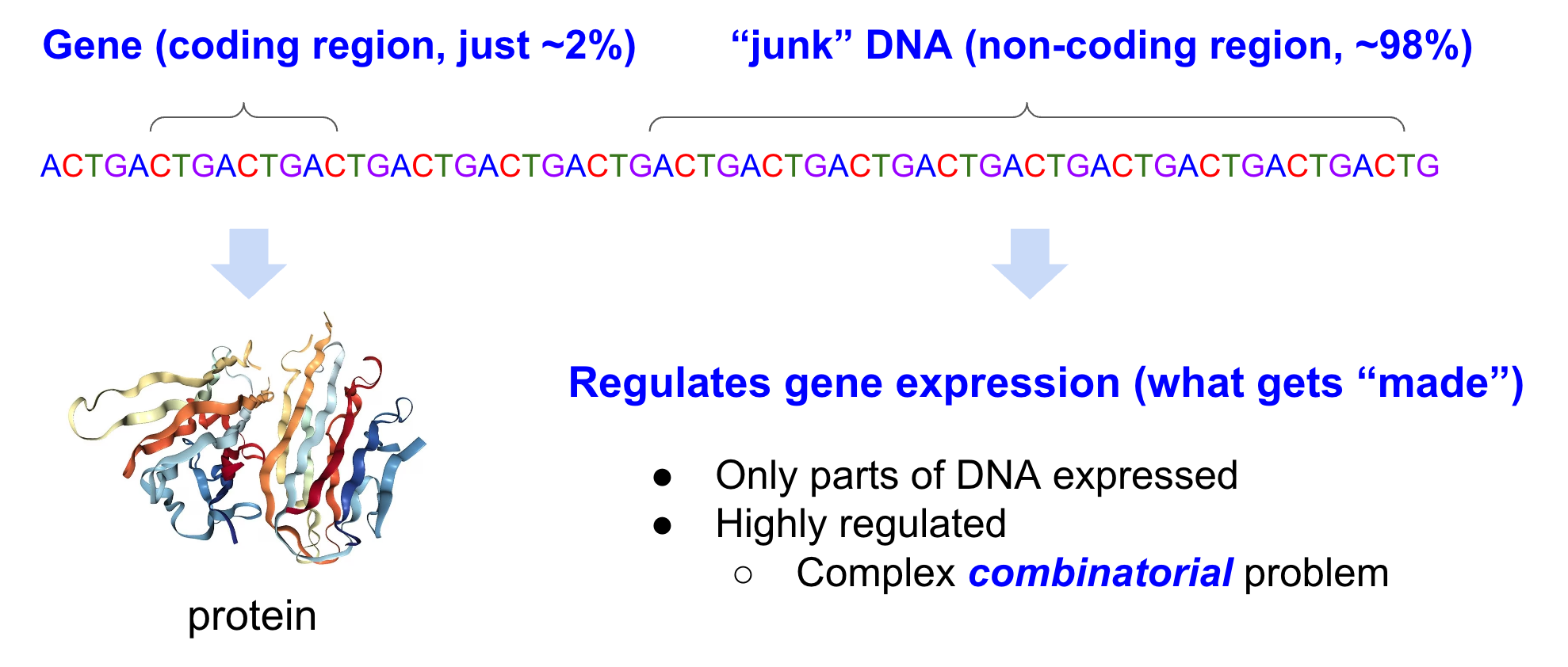
Genomic FMs can be thought of as modeling DNA as a complex combinatorial problem of, for example, DNA motifs that regulate gene expression and of protein coding regions.
In your DNA, there are coding regions, and non-coding regions. Coding regions, classically, encode instructions on how to make proteins. Previously, people thought non-coding regions were “junk” DNA, even though it accounts for 98% of the genome! We now know that non-coding regions regulate gene expression, and play a large role in cell differentiation. (For those CS-minded - think of gene expression as “instantiation”, with DNA a big template). Some believe there shouldn’t even be a distinction between coding and non-coding regions. Turns out understanding how gene expression is regulated is quite complex and very much an active research field.
How do the non-coding regions regulate gene expression physically? A protein complex called RNA polymerase needs to bind to DNA at specific sites to kick start gene expression. The presence of DNA motifs, or sequence patterns in non-coding regions, can alter the probability of whether RNA polymerase will bind. These motifs (aka regulatory regions) include promoters, enhancers, silencers, to name a few. Enhancers, in particular, can regulate the expression of their target genes that are hundreds of thousands of nucleotides away [Avsec et al.]. There are “first order” effects, ie, what motifs are present and where they are. In addition higher order effects, ie, a combination of these motifs, how they interact, what other proteins bind, all in turn also affect whether RNA polymerase will bind at a specific site.
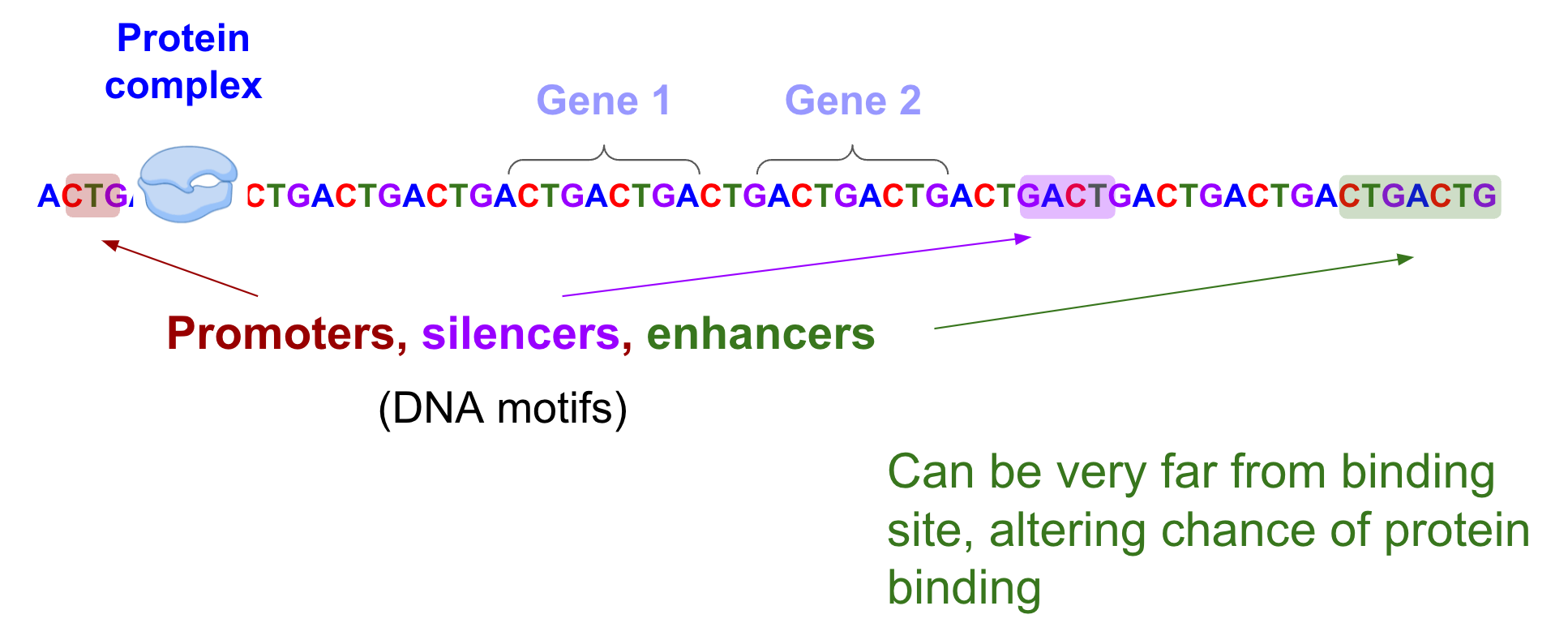
The protein complex (RNA polymerase) binds to DNA at specific sites to kick start gene expression. The presence of DNA motifs including promoters, silencers, and enhancers can have long-range interactions that regulate whether this binding occurs.
There are many other things that can effect gene expression (e.g. splices, epigenetics, transcription factors), but a big take-away is, we can model regulatory genomics as a complex combinatorial task of DNA motifs and their short and long-range interactions.
We know foundation models are pretty good at learning combinatorial representations! And Hyena is quite good at long-range interactions.
1M nucleotide-tokens with HyenaDNA
To model DNA and these interactions, HyenaDNA uses a simple stack of Hyena operators as its backbone (even more arch. details), and processes DNA sequences using a single character tokenizer. By learning the distribution of DNA sequences, HyenaDNA uses unsupervised learning to implicitly learn representations of how genes are encoded *and* how non-coding regions are involved in regulating gene expression.
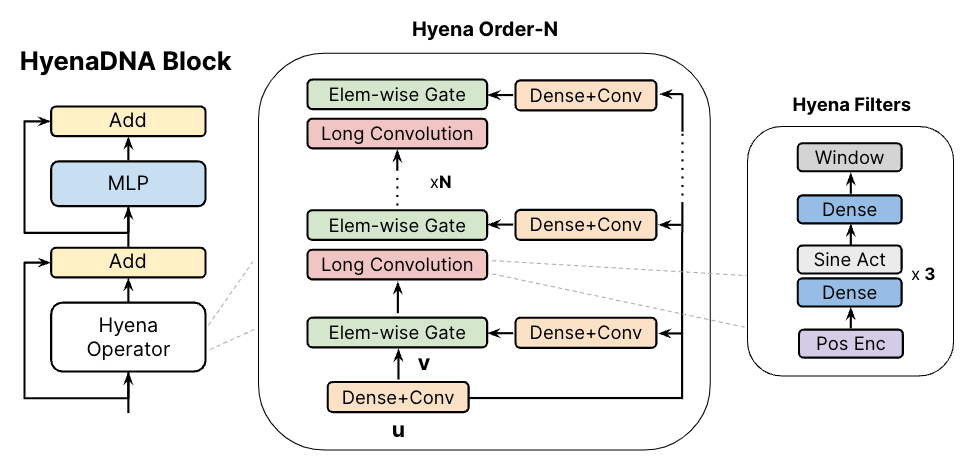
(left) The HyenaDNA block is similar to a Transformer-decoder block with the attention swapped for a Hyena Operator. (middle) The Hyena operator is an interleaving of long convolutions and element-wise gates. These gates use projections of the input, comprised of dense and short convolutional layers. (right) The long convolutions are implicitly parameterized via an MLP that creates the weights of long Hyena filters.
Technical highlights:
We train a family of HyenaDNA models with different sizes and context lengths, with a number of highlights we’re excited to share.
- Pretraining: context length of up to 1M tokens, 500x longer than previous genomic FMs
- Single nucleotide resolution and tokenization (a vocab of 4!), with global context at every layer
- Scales sub-quadratically in sequence length (N Log N) eg, trains 160x faster than Transformers at sequence length 1 million
- Introduce a sequence length warmup scheduler to address instability and to speedup training even more, eg, 40% reduction in training time on species classification at 450k nucleotides
- Parameter efficient:
It was important we have a model that was *accessible* to academics, and in particular, computational biologists (via model size, open source everything, simplicity). Check out the HuggingFace checkpoints :)
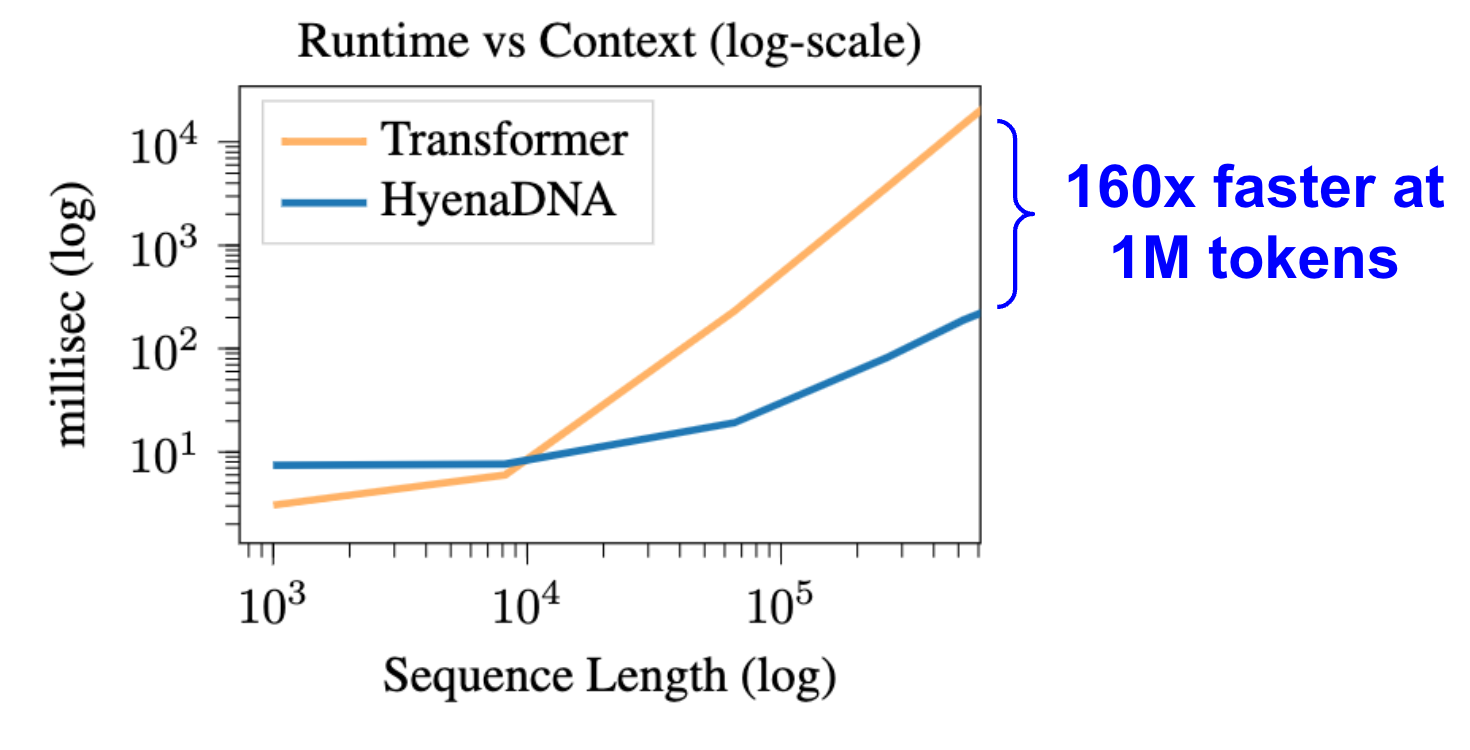
We benchmark forward and backward pass runtime for HyenaDNA vs Transformer (with Flash Attention). At 1 million tokens, HyenaDNA is 160x faster. Settings used: 1xA100 80GB, 2 layers, 128 model dimension, batch size 1, gradient checkpointing.
Pretraining on the Human Genome
For pretraining, we scale models varying model depth and width (i.e. size), and context length. We show that as context is increased, we’re able to improve perplexity (i.e. improve accuracy of next token prediction). On our longest sequence model, we reach 1 million tokens! Which is a 500x+ increase over previous dense-attention genomic FM models.
One thing we noticed was that with longer sequences at the nucleotide-character level (and smaller vocabulary), the ratio of model size and quality is far different than what we expected. HyenaDNA is a fraction of the size of previous genomic FMs, and we’re super excited about exploring this further (in other modalities too!).
So how does HyenaDNA work on actual genomic tasks?
Downstream tasks
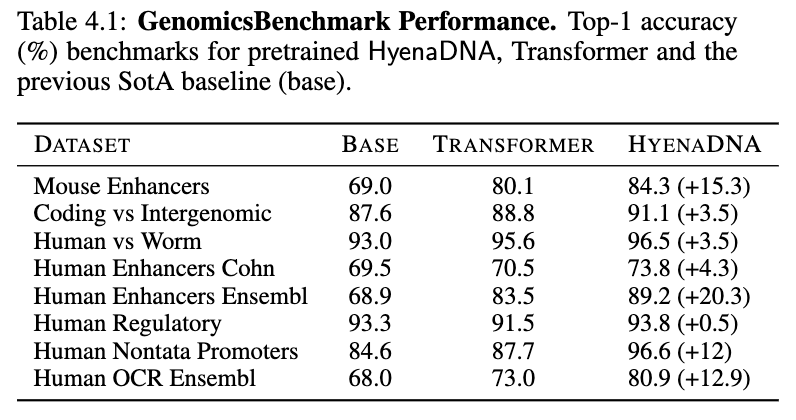
We apply our pretrained models on a number of downstream tasks, starting with “short" range tasks (< 5k tokens), to assess how well single nucleotide resolution works. On the GenomicBenchmarks dataset, we set new SotA on all 8 datasets, on average by 9 points and up to 20 points on enhancer prediction! See our sample HyenaDNA colab to try it out yourself!
On the benchmarks from the Nucleotide Transformer, we surpass SotA on 12 of 17 datasets, notably, with orders of magnitude less parameters and pretraining data1, as shown below.
Exploring in-context on genomics
In-context learning (ICL) has shown amazing capabilities in natural language, adapting to new data and tasks on the fly without standard fine-tuning. Recent work in our lab [TART] has started to close the gap between ICL and fine-tuning, but we noticed there were no baselines in ICL on genomics to begin with!
In our initial experiments, we noticed a big performance gap when using “pure” ICL (relying only on inference with few-shot demonstrations in the prompt) vs. standard fine-tuning. We hypothesize that the sequence structure in the DNA pretraining is far less diverse than in natural language, ie, no concept of labels or description follows a DNA sequence (whereas in natural language, there are many examples of how a class might follow a prompt). To even the playing field, we include a brief tuning phase to introduce HyenaDNA to the concept of classification using its existing limited vocabulary. This lets us use two variations of ICL methods that began to close the gap with standard fine-tuning.
Soft prompt Tuneable Tokens & Instruction Fine-Tuning
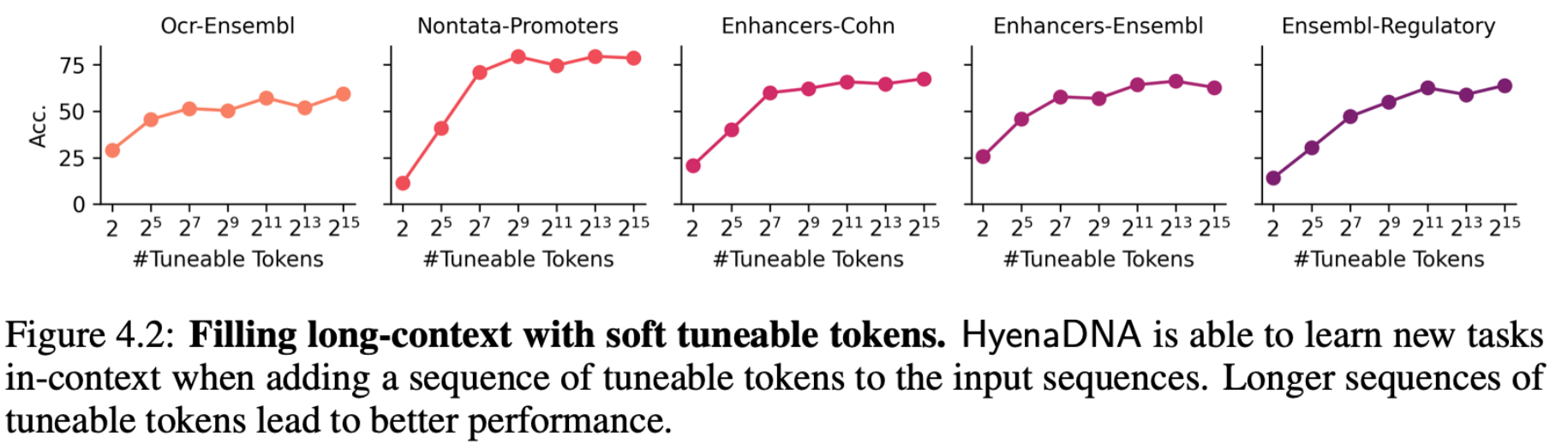
With long context opened up, we’re able to fill the input (prepend) with soft prompt tokens to guide the output of a frozen pretrained HyenaDNA model. The cool thing is that as we increase the number of soft prompt tokens (up to 32k), we improve accuracy reaching performance near the baselines on the GenomicBenchmarks datasets. No need to update the weights of the pretrained model or attach a decoder head!
Using just DNA sequences during pretraining, introducing the notion of a “class” for a classification task, for example, requires a clever approach. We repurpose the original DNA vocabulary (A, C, T, G) to indicate classes (e.g. “A” = True, “C” = False), and here's where we use a brief tuning phase to map the vocab to these classes.
In the second approach, we use instruction fine-tuning to stack demonstrations (a DNA sample and its class label, similar to instruction fine-tuning) in the input. Without a tuning phase ("pure" ICL), the model struggles. After a few "shots" of demonstration tuning the model does begin to learn (more details in the paper).
We’re excited about further work in genomics to further close the gap between standard fine-tuning and ICL!
Ultralong-range tasks
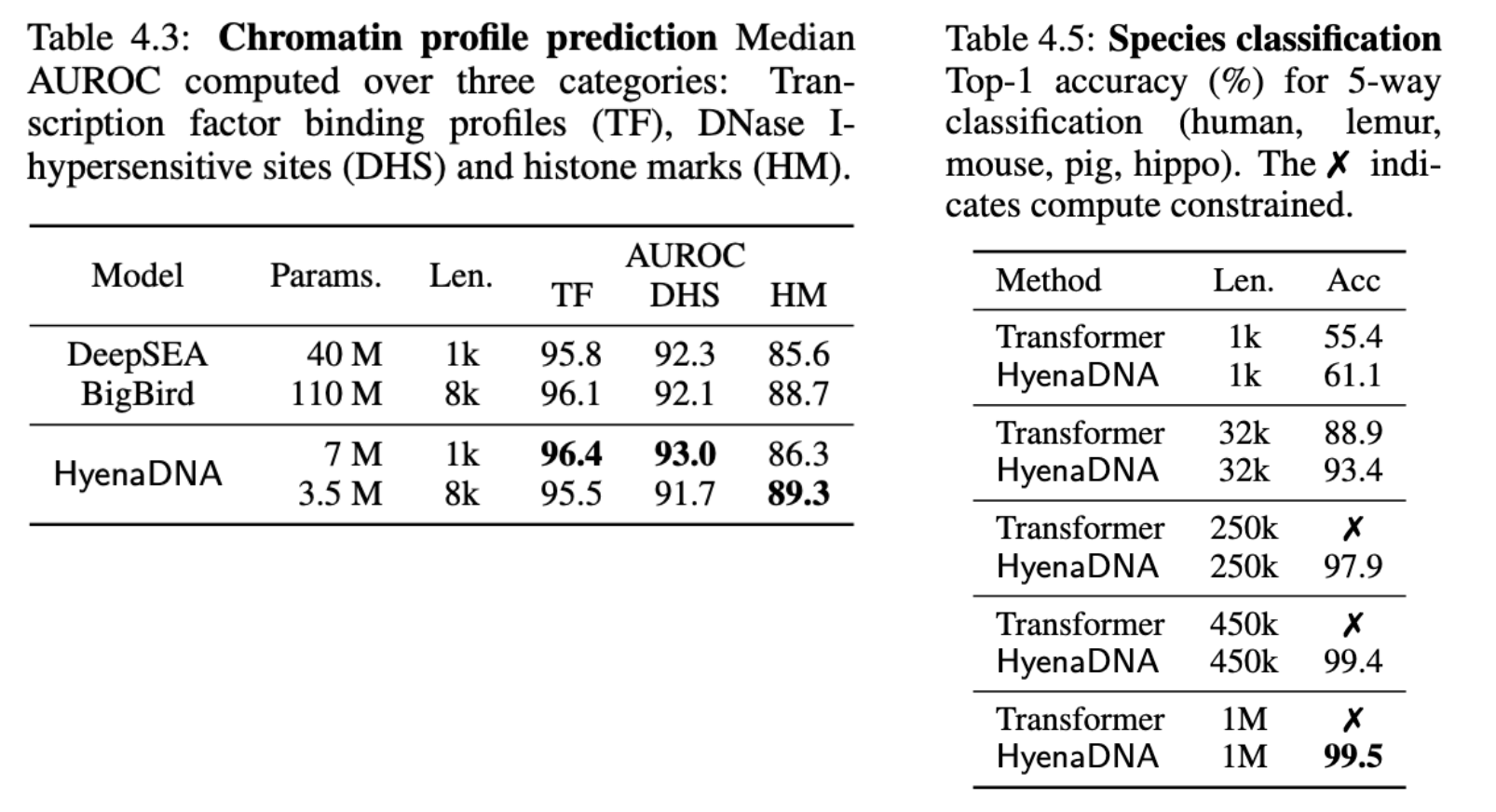
On a challenging chromatin profile task using a 919 multi-way task, HyenaDNA performs competively (with a far smaller model) against BigBird, a SotA sparse transformer. The tasks include simultaneously predicting the presence epigenetic markers, cleavage sites and transcription factor binding.
To isolate even longer range capabilities, we design a novel ultralong-range species classification, where we sample a random sequence from a set of 5 species. Since different species can share a lot of genetic code, increasing context length should enable a model to better differentiate between the species. Indeed, HyenaDNA is able to effectively solve the task by increasing context length to 450k and 1M tokens.
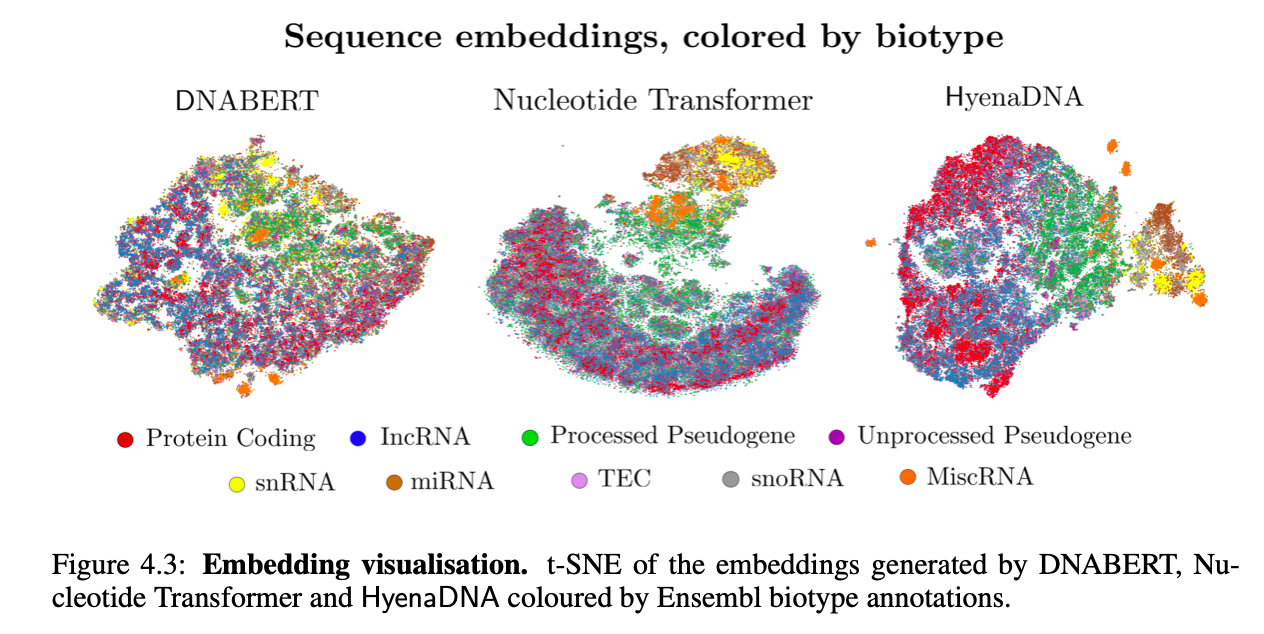
Visualizing Embedding Space
Finally, we visualize HyenaDNA pretrained embeddings against 2 popular genomic FMs: DNABERT and the Nucleotide Transformer. We show that HyenaDNA better separates between biotypes (genes and transcript function), notably with a much smaller model, by measuring F1 classification scores.
What's next
This is just the beginning - there are a lot of ideas we think Hyena/DNA could be applied to - DNA structure, protein complexes, generative and designs tasks!
But even better, we'd love to hear from folks about their high impact applications that could benefit from ultralong context. We're open to collaborators and to support where we can, so please reach out :)
Reaching a billion tokens would be fun, but pushing the limits of long context models for scientific & real-world impact - it doesn't get better than that!
Eric Nguyen, etnguyen@stanford.edu
Michael Poli, poli@stanford.edu
Marjan Faizi, faizi@berkeley.edu
We also have a Discord channel to help stir up ideas, tips, and advice on working with HyenaDNA on your projects :)
Update 10/15/23: we gave an extended talk (YouTube) at OpenBioML, where we breakdown how the Hyena operator works, and how we trained HyenaDNA.
Acknowledgments
This work would not have been possible without the entire HyenaDNA team!
Full author list: Eric Nguyen*, Michael Poli*, Marjan Faizi*, Armin Thomas, Callum Birch-Sykes, Michael Wornow, Aman Patel, Clayton Rabideau, Stefano Massaroli, Yoshua Bengio, Stefano Ermon, Stephen A. Baccus, Chris Ré
Also thanks to HAI, Google Cloud, Together, and SynTensor for donating compute.
*equal contribution
- On benchmarks from the Nucleotide Transformer, HyenaDNA uses a model with 1500x fewer parameters (2.5B vs 1.6M) and 3200 less pretraining data (3202 vs 1 human reference genome).↩