
Evo is a long-context biological foundation model that generalizes across the fundamental modalities of biology: DNA, RNA, and proteins. It is capable of both prediction tasks and generative design, from molecular to whole genome scale (generating 650k+ tokens). Evo is trained at a single-nucleotide, byte-level resolution, on a large corpus of prokaryotic and phage genomic sequences.
Resources
paper
repo
colab
together playground (browser tool)
API wrapper (by Nitro Bio)
huggingface (ckpts)
pip install
What can we learn from DNA?
We’ve long held the belief that biology is one of the primary domains that could truly benefit from long context models. Our first proof-of-concept, HyenaDNA, showed it could process sequences up to 1M tokens at the byte-level for predictive tasks. We were thrilled to see such positive reception and interest from entities in and out of academia [1,2,3,4,5,6]! As fun as this was, we knew we wanted to continue pushing capabilities of foundation models in biology.
So we set our sights on a much harder challenge: generative DNA design. The problem was, a good number of biologists asked, “what would you even do if you can design DNA?” We spent 5 months searching for a partner that shared the same vision, and that’s when we found the Arc Institute, where we joined forces to create a model called Evo. Our goal - to be able to generate an entire (prokaryotic) genome from scratch using AI.
Why? Understanding by creating was one motivation. But a real motivation that drove us here at Hazy Research, perhaps naively, was to create biomolecular machines using AI that can target cancer cells or any other disease we programmed it to. To create microbes for better biofuels, absorb carbon dioxide, or discover a new class of antibiotics. These were some of our dreams - and we didn’t succeed on any one of them.
And yet, we’ve never been more excited about the potential for generative DNA and biology.
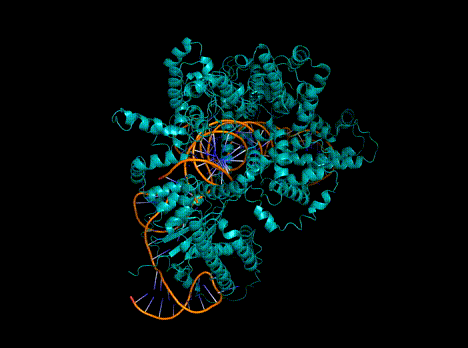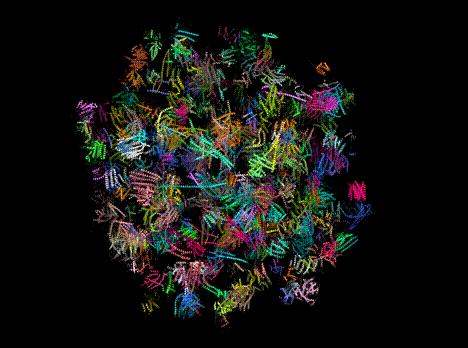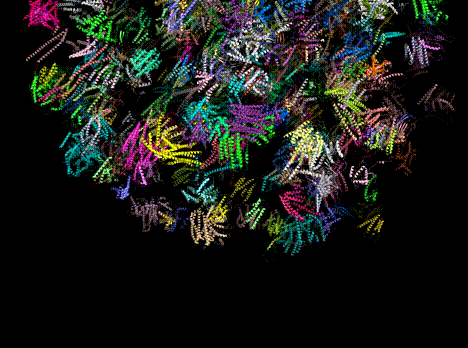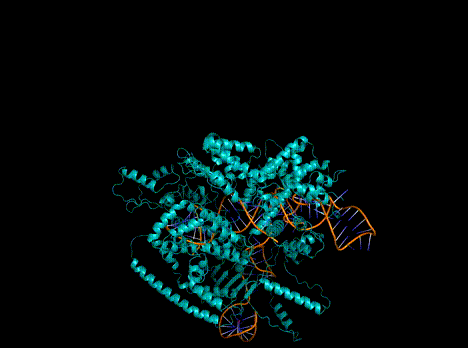
Figure 1. Predicted protein structures from a single, Evo-generated sequence.
As we were training Evo, compiling the largest DNA pretraining dataset (we know of), it felt like we were observing a “GPT” moment in biology. A simple unsupervised task was getting competitive zero-shot performance by modeling across the central dogma of biology, and generalizing across DNA, RNA and protein modalities.
One of the really exciting moments came when we were able to generate the world’s first AI designed CRISPR system (a complex of RNA and proteins), without requiring supervision. There were many other “firsts” that we observed throughout this project - but similar to natural language, there will be many more capabilities to emerge and yet to be discovered from the relatively untapped reservoir of biological data.
And so these are some of the reasons why we believe learning from DNA sequences is the next “grand challenge” of biology. If this blog doesn’t convince some folks in machine learning to work in biology, we will not have done our job!
It’s still very early days (think “blurry” GANs images), but the viability of scaling large biological foundation models is indeed promising. Let’s dive into the ML team "director's cut" on Evo.
Contents
Feel free to jump directly:
- Evo highlights
- challenges in modeling DNA
- DNA pretraining scaling laws
- Mechanistic Architecture Design
- CRISPR & gene essentiality results
- future directions
Evo highlights
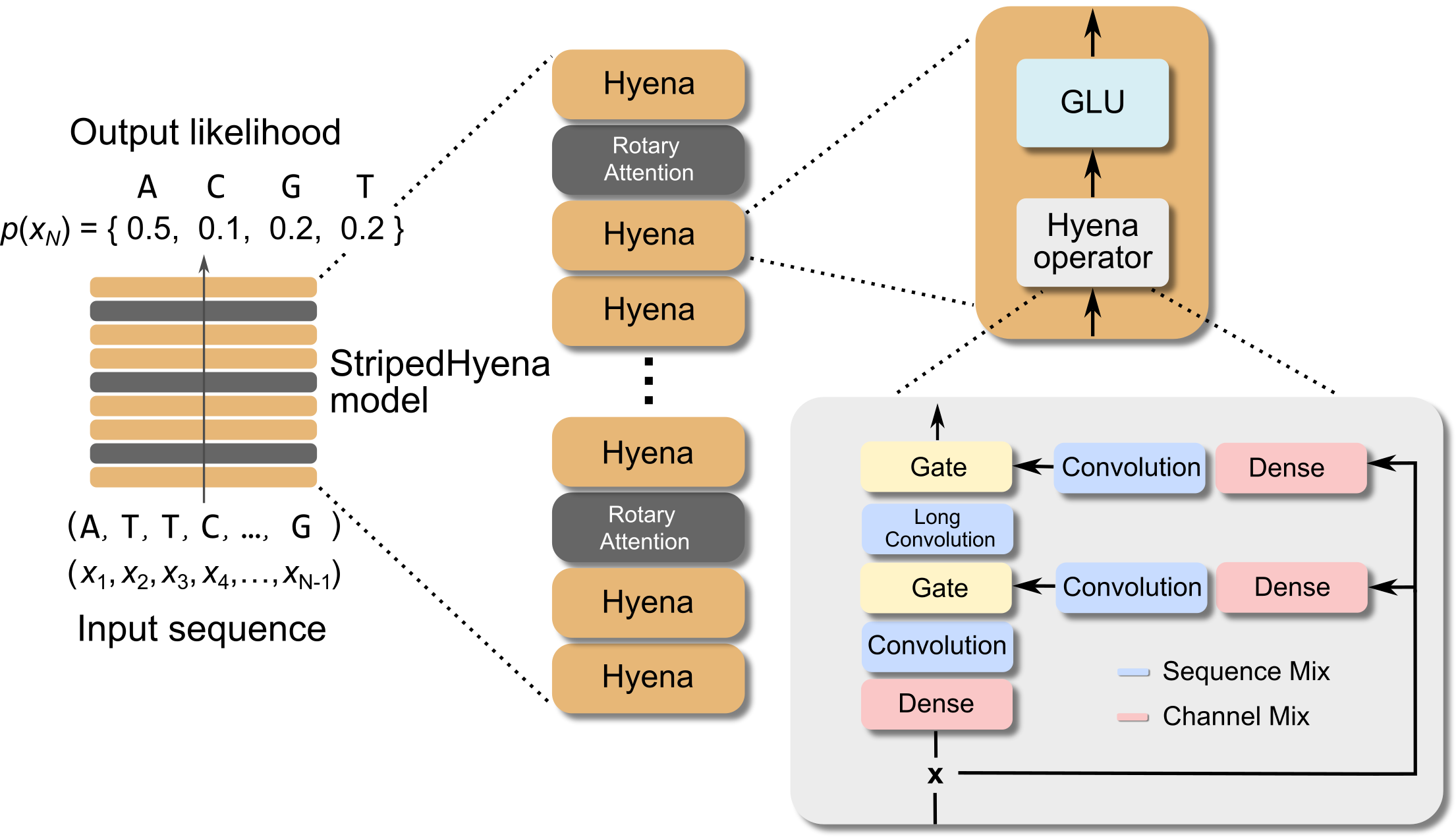
Figure 2. The Evo model architecture, based on StripedHyena.
With Hazy, Arc and Together AI, we trained Evo, a 7B parameter long context foundation model built to generate DNA sequences at single nucleotide resolution, and over 650k+ tokens long.
-
Evo is trained on 2.7M prokaryotic and phage genomes (un-annotated), using next token prediction. We compiled a dataset called OpenGenome, which we’re releasing open source, containing 300B tokens.
-
Evo is based on the StripedHyena architecture, an enhanced Hyena model that’s hybridized with rotary attention and trained in 2 stages using an efficient context extension, reaching a context of 131k tokens.
-
Beyond the technical innovations of the model (more in paper), the strength of the multimodal learning and generalization abilities of the model is what truly surprised us. Evo is able to learn across DNA, RNA and proteins, reaching competitive zero-shot performance on function prediction with state-of-the-art (SOTA) protein specific FMs like ESM2 and ProGen, notably, without explicitly being shown protein coding regions.
-
Evo understands at the whole genome level - using a gene essentiality test, Evo can predict which genes are essential to an organism’s survival based on small DNA mutations, also zero-shot (which currently is only possible in the wet lab).
-
Evo excels at generation, where we showcase the generation of molecules, to systems, and to whole genomes scale. One of the key breakthroughs we highlight, Evo can design novel CRISPR systems (including genes and RNA), an exciting frontier for creating new forms of genome editing tools.
We describe the biological innovations more in our initial blog release and in greater detail in the paper, so we’ll focus on what makes modeling DNA challenging and an exciting frontier for ML.
DNA as a language
Here, we largely treat DNA as a language, and so why haven’t we been able to effectively learn from DNA before? Aren’t Transformers great at language? This is something that initially blocked us as well. As we started scaling *thousands* of DNA models over the past year, several key lessons have emerged (obvious to many computational biologists).
DNA is not like natural language.
DNA is more like several languages or modalities in one. Typically, it’s believed that these biological languages (DNA, RNA, proteins) are so complex that they each require specialized models to effectively model them. But genomes carry an entire set of DNA to make a complete organism. And so it begs the question, “is DNA all you need”?
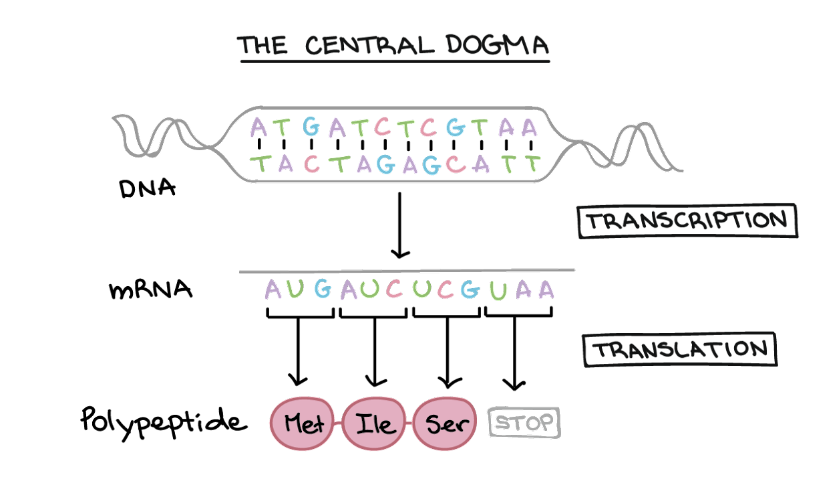
Figure 3. The fundamental “languages” of biology DNA, RNA, and proteins, united in the central dogma. (Photo credit: Khan Academy)
A key technical set of challenges in learning from DNA, among many, has been the long-range modeling (e.g. human genome is 3B nucleotides) as well as the resolution required to capture the effects of single mutations throughout evolution, which occur one nucleotide at a time. This can lead to varying “signal” strengths over the different modalities in DNA (discussed more in our first DNA blog). Though very long context models are emerging [Gemini, Claude], no large-scale Transformer has been effectively used at single character / byte level. This turns out to be an “achilles heel” in Transformers for DNA language modeling.
DNA scaling laws
To compare across potential architectures, we conducted the first DNA (and byte-level tokenization!) scaling laws experiments via the Chinchilla protocol. This involved training 300 models across prevailing language and DNA models: Transformer++, Mamba, Hyena, and StripedHyena, where we sweep across model size (6M to 1B) and dataset size, compute budgets and hyperparameters.
Scaling laws in natural language have helped propel LLM research forward by providing a guidebook on how to scale models and data with predictable performance. No such work has been done in DNA language modeling, and we hope to accelerate research in the field by releasing intermediate checkpoints during our training, hosted on HuggingFace. There’s a great deal of lessons learned that we hope to share with the community over time (perhaps a standalone blog?). There's so much we don't know about the best way to pretrain on DNA or with byte-level tokenization.
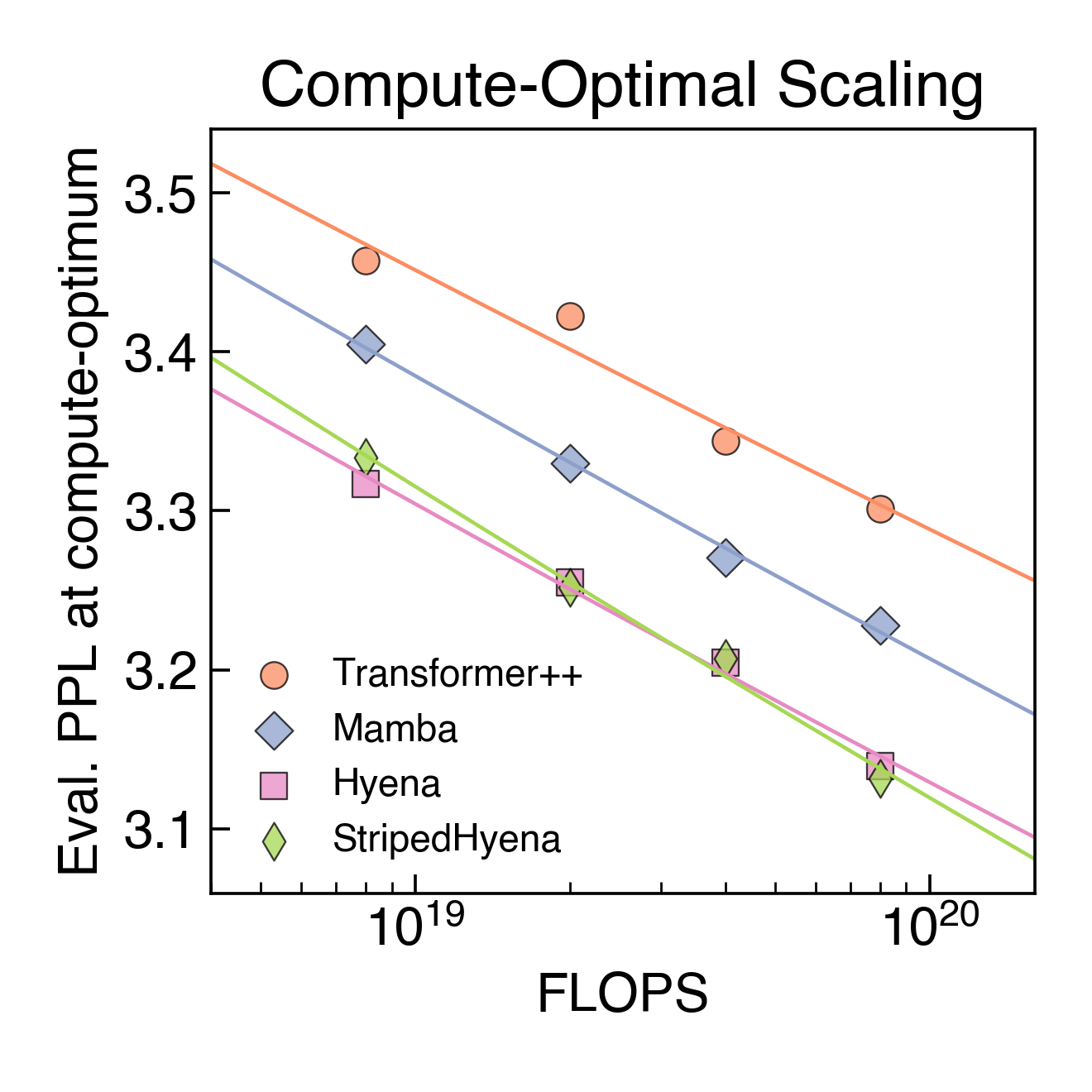
Figure 5. Compute optimal scaling on DNA pretraining. Each point represents the lowest perplexity for a model after sweeping different compute allocations (model size and training tokens).
In DNA scaling, we found Transformer++ to yield significantly worse perplexity (a measure of next token prediction quality) at all compute budgets, a symptom of the inefficiency of the architecture at the byte resolution [Charformer, CANINE]. State-space and deep signal processing architectures are observed to improve on the scaling rate over Transformer++, with Hyena and StripedHyena resulting in the best scaling rate. This suggests that at scale, the prevailing architecture in language may not easily transfer to DNA.
Mechanistic Architecture Design
In upcoming work on how we designed StripedHyena and other hybrid models, we dive deeper into understanding their advantages on DNA data by using a new framework we call Mechanistic Architecture Design (MAD). MAD builds on previous work in the lab [H3, Hyena, MQAR] in using targeted synthetic tasks that allowed us to test new architecture design choices. With MAD, we are able to show for the first time a connection between syntethic performance across various tasks and scaling laws.
These token manipulation tasks expand beyond previous recall-focused tasks, and include testing the ability to compress/aggregate multiple token representations (important in byte-level tokens) and to filter noise, where genomic sequences are known to contain a lot of “junk” DNA. Through synthetic tasks, we’re able to better understand where models do well or worse, and in turn, use this to drive the design of stronger models. Stay tuned.
Zero-shot prediction and multimodal generation
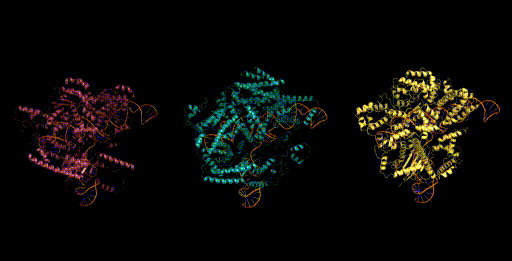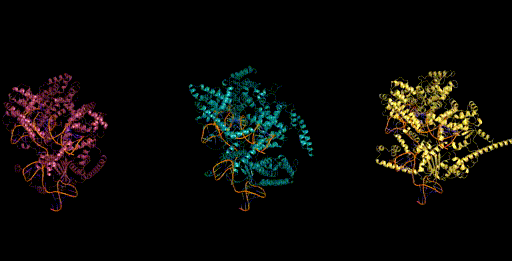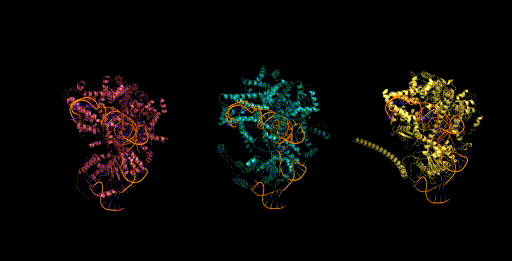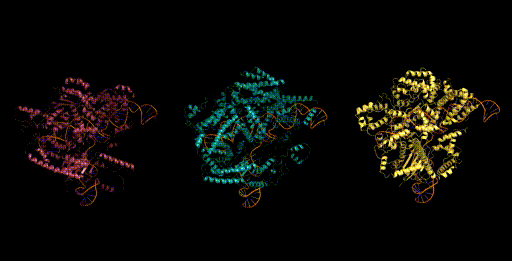
Figure 6. Generative design of CRISPR-Cas molecular complexes with Evo.
As a preview, we highlight one of the generative design capabilities of Evo. Today, generative models for biology usually focus on a single modality (at a time)—for example, only on proteins or on RNA. Instead, Evo can perform multimodal design to generate novel CRISPR systems, a task that requires creating large functional complexes of proteins and ncRNA (non-coding RNA), and is out of reach for existing generative models.
Typically, discovering new CRISPR systems requires searching through natural genomes for similar sequences that were literally taken from an organism. Promisingly, Evo enables a new approach to generating biological diversity by sampling sequences directly from a generative model, an exciting frontier for creating new forms of genome editing tools.
Strikingly, Evo can also understand biological function at the whole genome level. Using an in silico gene essentiality test on 56 whole-genomes (at 66 kbp), Evo can predict which genes are essential to an organism’s survival based on small DNA mutations. It can do so zero-shot and with no supervision. For comparison, a gene essentiality experiment in the laboratory could require 6 months to a year of experimental effort. In contrast, we replace this with a few forward passes through a neural network.
There are a range of capabilities (additional zero-shot predictions to generation of whole genome scale sequences) that we discuss in the initial blog and paper, and we encourage folks to check it out. But if that’s still too much time commitment for your comfort, we’re big fans of this summary video by Julia Bauman.
Future directions

We’re not working in a vacuum, there’s been a tremendous amount of fundamental research in biology (including computational, synthetic, bioengineering). Our goal is to accelerate the research where we can with the tools we know best, and hopefully motivating a few ML researchers along the way.
In the spirit of foundation models in other domains, we believe that Evo and other models that emerge can serve as a foundation for building useful things on top.
The potential to draw from the language modeling community is promising, as we incorporate prompt engineering and alignment techniques to design sequences with higher controllability and quality. Multimodal learning, and injecting domain specific knowledge are all things we’re bull-ish on.
While data and models are certainly important, evaluations play an outsized importance in biology, and perhaps is the biggest barrier to entry for the ML community. How do you formulate a task, what pretraining and annotations are needed, and of course, what does it mean in terms of the mechanistic understanding in biology? These are just some of the many questions we continue to think about as we try to innovate, as well as respect the body of work before us.
Even more challenging, how do we learn from eukaryotes (humans, mammals), which have far more complex genomes than prokaryotes. Is simply scaling one solution?
We’re in the early innings of what we think is possible in designing DNA sequences. Dropping perplexity on the Pile is exciting, and can lead to better downstream benchmarks or chatbots. Better perplexity on DNA and other bio sequences can lead to creating new tools for countless scientific advances and the improvement of human health.
We believe there's a big appetite in the ML community to work on meaningful problems (in bio or other) with cutting edge technology. If you've made it this far in the blog, you likely have that same hunger. Go after that grand challenge in biology, chemistry, climate tech, or whatever domain excites you. Tinker, collborate, and build the thing.
Correspondence
Eric Nguyen, etnguyen@stanford.edu
Michael Poli, polimic03@gmail.com
The full team also includes: Matt Durrant, Armin Thomas, Brian Kang, Jeremy Sullivan, Madelena Ng, Ashley Lewis, Aman Patel, Aarou Lou, Stefano Ermon, Stephen Baccus, Tina Hernandez-Boussard, Chris Ré, Patrick Hsu and Brian Hie.
Acknowledgements
We thank our labmates for their helpful feedback in writing this post: Jerry Liu, Michael Zhang, Jon Saad-Falcon, and Ben Viggiano.